Quick start¶
GPMap is a small Python package that subsets the pandas DataFrame to handle genotype-phenotype map data. The package include utilities to read/write data to/from disk, enumerate large sequence/genotype spaces efficiently, and compute various statistics from an arbitrary genotype-phenotype map.
GenotypePhenotypeMap object¶
The main object in gpmap is the GenotypePhenotypeMap object. The object stores data as a Pandas DataFrame, which can be accessed through the .data attribute. Your object will look something like this:
from gpmap import GenotypePhenotypeMap
# Data
wildtype = "AAA"
genotypes = ["AAA", "AAT", "ATA", "TAA", "ATT", "TAT", "TTA", "TTT"]
phenotypes = [0.1, 0.2, 0.2, 0.6, 0.4, 0.6, 1.0, 1.1]
stdeviations = [0.05, 0.05, 0.05, 0.05, 0.05, 0.05, 0.05, 0.05]
mutations = {
0: ["A", "T"],
1: ["A", "T"],
2: ["A", "T"]
}
# Initialize the object
gpm = GenotypePhenotypeMap(
wildtype,
genotypes,
phenotypes,
mutations=mutations,
stdeviations=stdeviations
)
# Check out the data.
gpm.data
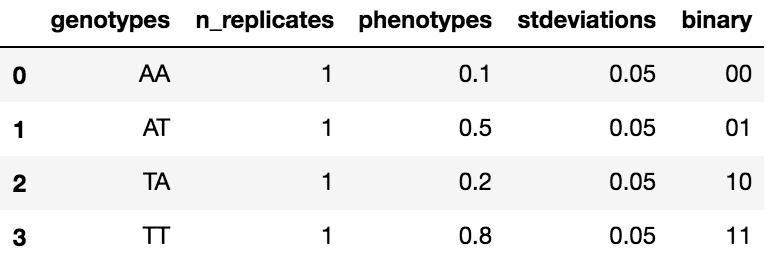
The underlying DataFrame will have at least 5 columns: genotypes, phenotypes, stdeviations, n_replicates, and binary. The binary column is computed by the GenotypePhenotypeMap object
mutations dictionary¶
The mutations dictionary tells GPMap what mutations, indels, etc. should be incorporated in the map. It is the most important data you pass to GPMap.
It is a regular Python dictionary and looks something like:
wildtype = "AA"
mutations = {
0: ["A", "B"],
1: ["A", "B"]
}
The key represents the position of site, and the value represents the states possible at each site. In this example, the sequences have two sites and each site is either “A” or “T”.
Non-changing sites
If a site doesn’t mutate, give it a value of None.
wildtype = "AAA"
mutations = {
0: ["A", "T"],
1: ["A", "T"],
2: None # This site does not change
}
Here, site 2 does not change. All sequences will only have an “A” at that site.
Indels
You can incorporate indels using the gap character:
wildtype = "AAAA"
mutations = {
0: ["A", "T"],
1: ["A", "T"],
2: None,
3: ["A", "-"] # Sometimes, this site doesn't exist.
}
Here, site 3 will toggle between an “A” and a missing residue “-” (deletion).
Port to NetworkX¶
In many cases, you might be interested in porting a GenotypePhenotypeMap to NetworkX. NetworkX provides powerful functions for analyzing and plotting complex graphs. We have written a separate package, named gpgraph, to easily port GenotypePhenotypeMap to NetworkX.